Agammaglobulinemia ligada al cromosoma X, lo crucial del diagnóstico y tratamiento oportunos
Agammaglobulinemia ligada al cromosoma X, lo crucial del diagnóstico y tratamiento oportunos |
INTRODUCCIÓN. a Agammaglobulinemia ligada al cromosoma X es un tipo de inmunodeficiencia primaria originada por una mutación en el gen que codifica a la proteína responsable del proceso madurativo de los linfocitos B, provocando la disminución o ausencia de inmunoglobulinas en sangre periférica y la predisposición a procesos infecciosos a repetición, sobre todo a nivel del tracto respiratorio y digestivo. La sospecha clínica orienta la solicitud de pruebas complementarias de forma secuencial. El tratamiento consiste en la administración sustitutiva de por vida de inmunoglobulina humana. CASO CLÍNICO. Se presentó el caso de un niño de 8 años de edad con infecciones respiratorias altas y bajas a repetición, con estudios radiográficos de tórax que revelaron una atelectasia persistente, en quien la sospecha clínica dio paso a los evaluativos inmunológico y genético. RESULTADOS. El diagnóstico fue realizado a los 6 años de edad con recuento sérico de inmunoglobulinas por debajo del rango para la edad, citometría de flujo con CD19+ del 0,08% y genética con mutación del gen BTK. Se instauró tratamiento con Inmunoglobulina humana a 400 mg/Kg cada 4 semanas, se monitorizó los niveles de IgG antes de cada infusión. DISCUSIÓN. La Agammaglobulinemia ligada al cromosoma X constituye una enfermedad poco prevalente e infradiagnosticada en la que la sospecha clínica representa la base del abordaje, lo que permitió el tratamiento sustitutivo apropiado. CONCLUSIÓN. El diagnóstico y tratamiento oportunos permitieron evitar el desarrollo de infecciones respiratorias graves, mejorar la calidad de vida del niño y el asesoramiento genético familiar.
Palabras Clave: Cromosoma X; Agammaglobulinemia; Linfocitos B; Infecciones del sistema respiratorio; Mutación; Salud del Hombre.
INTRODUCTION. X-linked Agammaglobulinemia is a type of primary immunodeficiency
caused by a mutation in the gene that encodes the protein responsible for the maturation
process of B lymphocytes, causing the decrease or absence of immunoglobulins in
peripheral blood and the predisposition to repeated infectious processes, especially
at the level of the respiratory and digestive tracts. Clinical suspicion guides the request
for complementary tests sequentially. The treatment consists of lifelong substitute
administration of human immunoglobulin. CASE REPORT. The case of an 8-year-old boy
with repeated high and low respiratory infections was presented, with chest radiographic
studies that revealed persistent atelectasis, in whom clinical suspicion gave way to
immunological and genetic evaluations. RESULTS. The diagnosis was made at 6 years of
age with serum immunoglobulin counts below the age range, flow cytometry with CD19 +
of 0,08% and genetics with BTK gene mutation. Treatment with human Immunoglobulin
at 400 mg / Kg every 4 weeks was initiated, IgG levels were monitored before each
infusion. DISCUSSION. X- linked Agammaglobulinemia is a rare and underdiagnosed
disease in which clinical suspicion represents the basis of the approach, which allowed
for appropriate replacement. CONCLUSION. Timely diagnosis and treatment allowed to
avoid the development of serious respiratory infections, improve de child ́s quality of life
and family genetic couseling.
Keywords: X Chromosome; Agammaglobulinemia; B – lymphocytes; Respiratory Tract Infections; Mutation; Men ́s Health.
La Agammaglobulinemia ligada al cromosoma X (XLA) o Enfermedad de Bruton, es un tipo de inmunodeficiencia primaria humoral, congénita y hereditaria, que afecta a varones y a 1 de cada 200.000 nacidos vivos. Es provocada por una mutación en el gen BTK localizado en el brazo largo del cromosoma X (Xq21.3- Xq22) que codifica a la proteína Tirosin kinasa de Bruton que interviene en el proceso madurativo de los linfocitos B, dando lugar a fallos en las etapas tempranas del desarrollo de estas células, mientras que, la función de las células T se encuentra conservada1-5. Figura 1.

Esta inmunodeficiencia se caracteriza por la ausencia o reducción marcada en el recuento de células B, con la consecuente disminución o ausencia de todos los isotipos de inmunoglobulinas (Ig) en sangre periférica y el pobre desarrollo de los órganos linfoides (amígdalas, bazo, placas de Peyer y ganglios linfáticos). Se manifiesta con infecciones bacterianas recurrentes a nivel respiratorio y digestivo que se presentan generalmente a partir del sexto mes de vida, momento en que los anticuerpos maternos transferidos por vía placentaria empiezan a declinar2,4-7.
A nivel respiratorio las manifestaciones pueden ser de tipo infeccioso y no infeccioso. Dentro del primer grupo prevalecen la otitis media, rinosinusitis, bronquitis, y neumonía, generadas predominantemente por bacterias encapsuladas (Streptococcus pneumoniae, Haemophilus influenzae y Staphylococcus aureus) y en ocasiones por gérmenes oportunistas, con la característica de que el curso evolutivo es severo, persistente y recurrente. La presentación respiratoria puede incluir además complicaciones no infecciosas como bronquiectasias, engrosamiento de la pared bronquial, Enfermedad Pulmonar Intersticial, que son el resultado de las infecciones recurrentes y no controladas. Por otro lado, infecciones gastrointestinales provocadas por Campylobacter jejuni y Giardia lamblia son frecuentemente encontradas. Mientras que un tercio de los pacientes pueden debutar con infecciones graves como sepsis, meningitis, osteomielitis, artritis séptica, empiema, o celulitis2,8-12.
Alrededor del 50,0% de pacientes tienen el antecedente de un miembro de su familia afecto por la enfermedad; sin embargo, en un 15,0% se han identificado mutaciones de novo7,10.
Este caso tuvo como objetivo describir las características de una enfermedad poco prevalente, recalcando la importancia de la sospecha clínica como origen del diagnóstico y tratamiento oportuno.
Escolar masculino de 8 años de edad, segundo hijo de una pareja no consanguínea, sin antecedentes familiares de inmunodeficiencias, nace a término por cesárea, sin complicaciones, antropometría referida como normal, crecimiento y desarrollo adecuado e inmunizaciones completas. A partir del tercer año de vida presentó infecciones respiratorias altas a repetición (otitis y sinusitis), con historial de adenoidectomía y colocación de tubos de timpanostomía.
A los 5 años de edad, tuvo el primer episodio de neumonía tratada de forma ambulatoria. A los 6 años 5 meses de edad se realizó Sinusotomías maxilares y Timpanostomías con drenaje bilateral, dos meses después, presentó cuadro de Neumonía bacteriana que ameritó manejo hospitalario y rotación de tratamiento antibiótico durante su evolución. A los 6 años 8 meses de edad, se realizó la segunda hospitalización por diagnóstico de Neumonía más atelectasia derecha.
A los 6 años 9 meses de edad se inició el abordaje diagnóstico ante sospecha clínica de inmunodeficiencia primaria. El laboratorio registró en el recuento sérico hemograma normal y reactantes de fase aguda negativos. La radiografía de tórax reveló: atelectasia en base pulmonar derecha. Figura 2. En la TC de tórax se observó consolidación en el segmento 6 derecho, con broncograma aéreo, bronquiectasias circundantes, y engrosamiento de la pared de los bronquios. Figura 3.

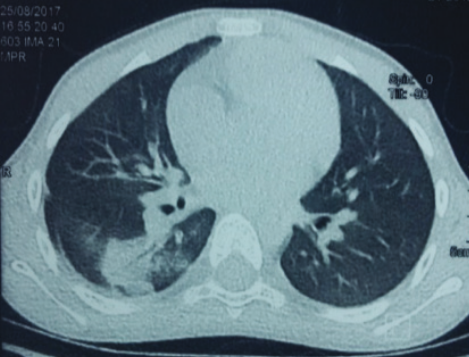
En el cultivo de esputo se reportó Enterobacter cloacae complex y Klebsiella oxytoca. Baciloscopía negativa. En la broncoscopia con lavado bronquial se observó abundante cantidad de moco blanquesino espeso en árbol bronquial derecho y ausencia visible de cuerpo extraño.
Los niveles de inmunoglobulinas por Nefelometría fueron: IgG 98 (751 – 1560), IgM 14 (46 – 304), IgA 6 (82 – 453 mg/dL), IgE < 1 (3-9 años: < 52 IU/mL). CD4: 946, CD8: 1840, Ratio 0.51. Citometría de flujo: CD19+: 2,15 (0,08%), CD3+: 2419,44 (93,79%), CD16+CD56+: 117,75 (4,56%) y, estudio de 10 mutaciones para Fibrosis Quística negativo. En el estudio genético por secuenciación de nueva generación se registró una variante patológica en el gen BTK. Exon 16, c.1581_1584delTTTG (p.Cys527Trpfs*2).
Se instauró tratamiento con Inmunoglobulina humana a 400 mg/kg intravenoso cada 4 semanas, con el objetivo de mantener valores preinfusionales sobre 500 mg/dL; sin embargo, al séptimo mes de tratamiento, con IgG 477 mg/dL, se decidió el incremento de dosis a 500 mg/kg, y se logró mantener al paciente libre de infecciones respiratorias. Al noveno mes de tratamiento presentó un cultivo de esputo con desarrollo de Aspergillus fumigatus, para lo que recibió tratamiento específico. Figura 4.
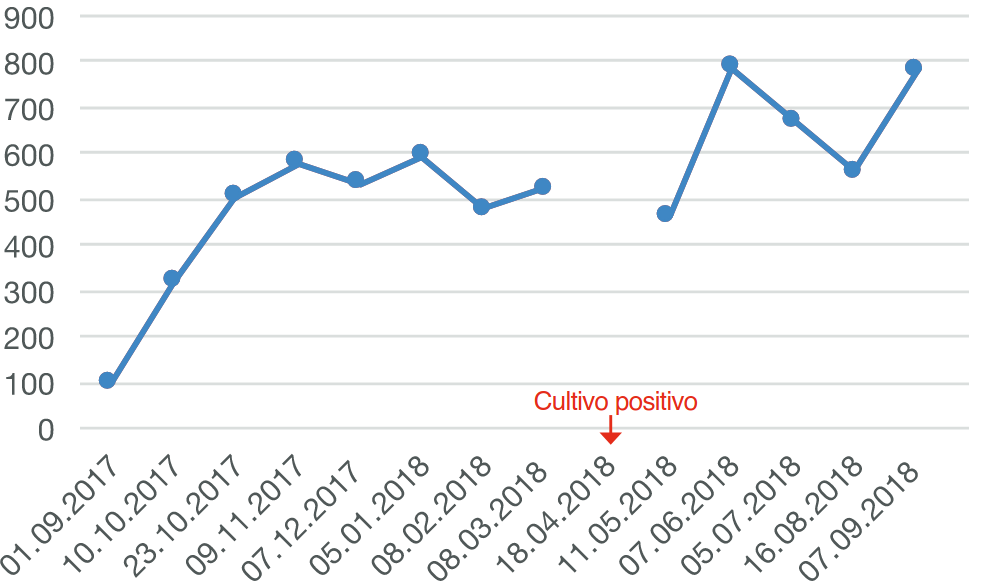
Al décimo sexto mes de tratamiento, por falta de medicación, se suspendió la administración de Ig por un lapso de 8 semanas, llevando a una caída en el nivel sérico de IgG a 265 y el consecuente desarrollo de infecciones respiratorias, presentando cuadro de Neumonía y Pansinusitis. Figura 5.

Al reiniciar el tratamiento se administró una dosis de 600 mg/Kg y se acortó el intervalo de administración cada tres semanas por dos ocasiones, posteriormente se regresó al esquema inicial con dosis 500 mg/kg cada 4 semanas, con lo que se logró mantener valores séricos sobre 700 mg/dL y el control de la sintomatología respiratoria.
Además se dio asesoramiento genético a la madre, indicando la importancia de realizar examen de pesquisa a ella y sus dos hijas mujeres para conocer si son portadoras de la enfermedad y establecer el riesgo de recurrencia; sin embargo, no fue posible la realización del estudio genético familiar por su costo.
Los pacientes con Agammaglobulinemia ligada al cromosoma X, cursan con infecciones bacterianas a repetición sobre todo a nivel del tracto respiratorio alto (75,0%), seguidas del tracto respiratorio bajo (65,0%), el tracto gastrointestinal (35,0%), la piel (28,0%) y el sistema nervioso central (16,0%)2,11,13.
Al examen físico, las amígdalas, adenoides y ganglios linfáticos cervicales suelen ser extremadamente pequeños, condición que debería alertar a los médicos2,7,10.
Los estudios complementarios deben ser solicitados en orden secuencial, partiendo de determinaciones de primera línea como: hemograma completo con recuento celular, reactantes de fase aguda, análisis de orina, cultivos y pruebas de imagen. La radiografía de cavum puede revelar la ausencia de tejido adenoideo, mientras que, la radiografía de tórax puede mostrar hallazgos en relación a infección pulmonar como atelectasias, engrosamiento de la pared bronquial o bronquiectasias. En la tomografía se puede observar a nivel de los senos paranasales hallazgos de sinusitis crónica, mientras que a nivel pulmonar la presencia de daño pulmonar y cuantificar la extensión del mismo2,9,11,14.
El abordaje continúa con la solicitud del recuento de inmunoglobulinas en suero, cuya interpretación deberá tener en cuenta los valores normales para la edad. La mayoría de pacientes con XLA muestran niveles séricos de Inmunoglobulina G menores a 200 mg/dL, con niveles de Ig A e Ig M menores a 20 mg/dL. Posteriormente se debe realizar el recuento de subpoblaciones linfocitarias (T, B y NK) mediante citometría de flujo la cual revelará un número reducido de linfocitos B, generalmente menor al 1,0%, y para el diagnóstico definitivo se requiere de la detección de mutaciones en el gen BTK, de las que han sido descritas alrededor de 8002,10,15–18.
La Sociedad Europea de Inmunodeficiencias ha establecido los criterios diagnósticos para considerar un caso definitivo, probable y posible18. Tabla 1.
El tratamiento consiste en la administración sustitutiva de por vida de Inmunoglobulina humana, en una dosis de 400 – 600 mg/Kg; sea por vía intravenosa o subcutánea, en el primer caso con un intervalo de 2 – 4 semanas, mientras que en el segundo cada 1 a 14 días, con el objetivo de lograr valores preinfusionales de Ig G entre 500 - 800 mg/ dL; además del tratamiento antibiótico adecuado para cualquier infección sospechada o documentada2,3,18,19.
La terapia con inmunoglobulina disminuye la incidencia y la severidad de las infecciones, el número de ingresos hospitalarios, la morbilidad por complicaciones crónicas y aumenta la esperanza de vida9,17,20.
La pesquisa familiar para la detección de portadoras y el diagnóstico prenatal son piezas claves en el manejo de las familias con este tipo de inmunodeficiencia. Se debe tener en consideración que, por tratarse de un patrón de herencia recesiva ligada al sexo, las mujeres son portadoras, mientras que los varones al tener un solo cromosoma X desarrollan la enfermedad si el gen está afectado. Cuando el paciente tiene antecedentes familiares de inmunodeficiencia en tíos o primos maternos, la madre es portadora de XLA, y, las hermanas y tías del paciente tienen un riesgo del 50,0% de ser portadoras. Ellas transmiten el carácter al 50,0% de sus hijas, quienes también serán portadoras, y al 50,0% de sus hijos quienes serán enfermos. Pero cuando el paciente no presenta una aparente historia familiar de inmunodeficiencia, alrededor del 90,0% de varones con laboratorio sugestivo tienen una mutación en el gen BTK10,21.
Caso definitivo: |
Caso probable: |
Caso posible: |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Fuente. Seoane M y Múñoz M (2006)18. Elaborado por. Autoras | ||||||||||
Varón con menos del 2,0% de
células B CD19+ y al menos una
de las siguientes premisas: |
Varón con menos del 2,0%
de células B CD19+, y
las siguientes premisas sean
positivas: |
Varón con menos del 2,0% de
células B CD19+, en el cual
estén excluidas otras causas de
hipogammaglobulinemia y tenga
al menos uno de lo siguiente: |
||||||||
- Mutación en BTK |
- Infecciones bacterianas
recurrentes en los primeros 5
años de vida. |
- Infecciones bacterianas
recurrentes en los primeros 5
años de vida |
||||||||
- Ausencia de ARN mensajero
BTK mediante análisis en
neutrófilos o monocitos |
- Niveles de IgG, IgA e IgM
más de 2 DE por debajo de los
valores normales para la edad |
- Niveles de IgG, IgA e IgM
más de 2 DE por debajo de los
valores normales para la edad |
||||||||
- Ausencia de proteína BTK en
monocitos o plaquetas |
- Ausencia de isohemaglutininas
y/o escasa respuesta en la
producción de anticuerpos frente
a vacunas |
- Ausencia de isohemaglutininas |
||||||||
- Primos, tíos o sobrinos
maternos con menos del 2,0% de
células B CD19+ |
- Exclusión de otras causas de
hipogammaglobulinemia. |
|||||||||
.
La Agammaglobulinemia ligada al cromosoma X constituye una enfermedad poco prevalente, infradiagnosticada o diagnosticada de forma tardía, que debe ser sospechada en varones con infecciones a repetición del tracto respiratorio y aparato digestivo.
La Historia Clínica Única, los antecedentes familiares y el examen físico detallado orientan la solicitud de pruebas de laboratorio, para lograr el diagnóstico oportuno y la instauración del tratamiento sustitutivo con Inmunoglobulina, lo que evita el deterioro de los órganos por las infecciones continuas y recurrentes así como la reducción de la morbi-mortalidad asociada, permite mejorar la calidad de vida de estos pacientes y brindar el asesoramiento familiar para la detección de portadoras.
XLA: Agammaglobulinemia ligada al cromosoma X; BTK: Tirosin kinasa de Bruton; Ig: Inmunoglobulina.
CV: Concepción y diseño del trabajo. Redacción del manuscrito. CV, JH: Recolección de información; revisión crítica del manuscrito, aprobación de su versión final. Todas las autoras leyeron y aprobaron la versión final del artículo.
Carla Lucía Vaca Yépez. Medico. Universidad Central del Ecuador. Especialista en Pediatría, Pontificia Universidad Católica del Ecuador. Servicio de Pediatría. Centro Clínico Quirúrgico Ambulatorio Hospital del Día Sangolquí. Sangolquí-Ecuador. ORCID ID: https://orcid.org/0000-0003-3168-9501
Jaira Lorena Hidalgo Vásconez. Medica General, Universidad Nacional de Loja. Especialista en Pediatría, Universidad Internacional del Ecuador. Médico Pediatra. Unidad Técnica de Pediatría, Hospital de Especialidades Carlos Andrade Marín. Quito-Ecuador. ORCID ID: https://orcid.org/0000-0001-8127-6137
Fueron utilizados recursos bibliográficos de uso libre y limitado. La información recolectada está disponible bajo requisición al autor principal.
El artículo científico fue aprobado por pares y por el Comité de Ética de Investigación en Seres Humanos – CEISH/ HECAM.
La publicación fue aprobada por el Consejo Editorial del HECAM.
El estudio fue financiado por las autoras.
Los autores reportaron no tener ningún conflicto de intereses personal, financiero, intelectual, económico y de interés corporativo.