Perfil clínico y epidemiológico de los pacientes con diagnóstico de neuromielitis óptica.
Perfil clínico y epidemiológico de los pacientes con diagnóstico de neuromielitis óptica. |
Perfil clínico y epidemiológico de los pacientes con diagnóstico de neuromielitis óptica.
Clinical and epidemiological profile of patients with neuromyelitis optica diagnosis.
Edgar Patricio Correa Díaz1, Francisco José Caiza Zambrano1, Wilson Alfredo Gualotuña Pachacama2, Joselyn Elizabeth Miño Zambrano2.
INTRODUCCIÓN. El trastorno del espectro de neuromielitis óptica, enfermedad inflamatoria, desmielinizante, afecta al sistema nervioso central, frecuente en poblaciones no caucásicas como la ecuatoriana. El retraso en su diagnóstico y tratamiento provoca discapacidad que se puede prevenir. OBJETIVO. Determinar el perfil clínico y epidemiológico de los pacientes con diagnóstico de trastorno del espectro de neuromielitis óptica. MATERIALES Y MÉTODOS. Estudio descriptivo transversal. Población de 45 Historias Clínicas y una muestra de 41 de pacientes con diagnóstico de trastorno del espectro de neuromielitis óptica atendidos en la Unidad de Neurología del Hospital de Especialidades Carlos Andrade Marín, período enero 2005 a diciembre 2019. Se realizó análisis univarial. Se aplicó el programa estadístico International Business Machines Statistical Package for the Social Sciences, versión 25. RESULTADOS. El 76,0% (31; 41) fueron mujeres. Datos promedios: edad 48,9 años; diagnóstico definitivo demoró 4,12 años, desde el inicio de los síntomas; tiempo de diagnóstico fue 3,17 años; 3,7 brotes en total; el 87,8% (36; 41) con un fenotipo recurrente. La media de duración de la enfermedad fue de 6,8 años. En el 70,7% (29; 41), se identificaron anticuerpos anti-AQP4 en suero mediante inmunofluorescencia directa, el 51,2% requirieron para la marcha apoyo uni o bilateral. El 43,9% (18; 41) debutó con neuritis óptica; el 31,7% (13; 41) presentaron mielitis como primer síntoma y el 24,4% (10; 41) la combinación de neuritis óptica y mielitis fueron los síntomas iniciales. CONCLUSIÓN. Se determinó el perfil clínico y epidemiológico de los pacientes con diagnóstico de trastorno del espectro de neuromielitis óptica. Existió demora en el diagnóstico definitivo de los pacientes desde el inicio de los síntomas, lo que se tradujo en un aumento de la discapacidad.
Palabras clave: Neuromielitis Óptica; Mielitis; Neuritis Óptica; Sistema Nervioso; Enfermedades Autoinmunes; Salud de la Persona con Discapacidad.
INTRODUCTION. Neuromyelitis optica spectrum disorder, an inflammatory, demyelinating disease, affects the central nervous system, common in non-Caucasian populations such as Ecuadorians. The delay in its diagnosis and treatment causes disability that can be prevented. OBJECTIVE. To determine the clinical and epidemiological profile of patients diagnosed with neuromyelitis optica spectrum disorder. MATERIALS AND METHODS. Cross-sectional descriptive study. Population of 45 Medical Records and a sample of 41 patients with a diagnosis of neuromyelitis optica spectrum disorder seen at the Neurology Unit of the Carlos Andrade Marín Specialties Hospital, period from January 2005 to December 2019. Univariate analysis was performed. The statistical program International Business Machines Statistical Package for the Social Sciences, version 25 was used. RESULTS. 76,0% (31; 41) were women. Average data: age 48,9 years; definitive diagnosis took 4,12 years from the onset of symptoms; time to diagnosis was 3,17 years; 3,7 outbreaks in total; 87,8% (36; 41) with a recurrent phenotype. The average disease duration was 6,8 years. In 70,7% (29; 41), anti-AQP4 antibodies were identified in serum by direct immunofluorescence, 51,2% required uni- or bilateral support for walking. Optic neuritis started in 43,9% (18; 41); 31,7% (13; 41) had myelitis as the first symptom and 24,4% (10; 41) the combination of optic neuritis and myelitis were the initial symptoms. CONCLUSION. The clinical and epidemiological profile of patients diagnosed with neuromyelitis optica spectrum disorder was determined. There was delay in the conclusive diagnosis of patients from the beginning of symptoms, which resulted in increased disability.
Keywords: Neuromyelitis Optica; Myelitis; Optic Neuritis; Central Nervous System; Autoinmune Diseases; Health of the Disabled.
El trastorno del espectro de neuromielitis óptica (NMO) es una enfermedad inflamatoria, autoinmune y desmielinizante que puede afectar diferentes áreas del sistema nervioso central (SNC) que provoca déficits importantes y permanentes en los pacientes1,2.
Eugene Devic fue el primero en describir la enfermedad en 1894 y desde 1936 se la asoció como una variante de la esclerosis múltiple (EM). Sin embargo, la identificación de la acuaporina 4 (AQP4) como un blanco inmunogénico en el año 2004, permitió una mejor comprensión de la fisiopatología de la enfermedad. Los hallazgos de resonancia magnética, mecanismos patológicos y sobre todo inmunológicos establecen claras diferencias entre estas dos entidades3,4.
La enfermedad es más frecuente en poblaciones no caucásicas donde la prevalencia de EM es baja, como el Ecuador, aunque no se conoce la prevalencia exacta de la enfermedad; y son limitados los datos en Latinoamérica5,6.
El objetivo de este estudio fue determinar el perfil clínico y epidemiológico de los pacientes con diagnóstico de trastorno del espectro de NMO que fueron atendidos en la Unidad de Neurología del Hospital de Especialidades Carlos Andrade Marín.
Estudio descriptivo de corte transversal. Población de 45 Historias Clínicas del Sistema AS400 y muestra de 41 sobre pacientes con diagnóstico de NMO, de la Clasificación Internacional de Enfermedades CIE-10-G36.0, atendidos en la Unidad de Neurología del Hospital de Especialidades Carlos Andrade Marín, periodo enero 2005 a diciembre 2019. Se incluyó datos confidenciales, codificados de ambos sexos, mayores de 18 años que cumplieron los criterios diagnósticos para NMO 20157. Se excluyó a cuatro pacientes por no cumplir con los criterios establecidos. Se consideraron variables sociodemográficas, de la enfermedad y del tratamiento. Se registraron datos de la escala expandida de discapacidad (EDSS, por sus siglas en inglés). Se realizó análisis univariado para la descripción de características generales de la población y de la enfermedad. Para la tabulación y análisis de datos se utilizó el International Business Machines Statistical Package for the Social Sciences (IBM SPSS), versión 25.
La caracterización demográfica determinó un 76,0% (31; 41) fueron mujeres, con una relación 3:1, autoidentificados como mestizos el 100,0%. El promedio de edad fue 48,9 años (DE +/- 14). La distribución de casos por provincias, según su lugar de nacimiento se muestra en la Tabla 1.
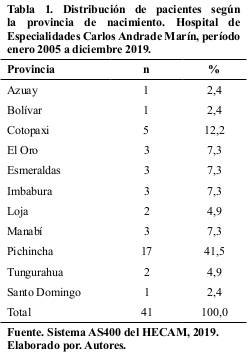
El 39,0% (16; 41) tuvo instrucción superior con un promedio de 11,6 (DE +/- 3,65) años de estudio y el 65,9% (27; 41) mantenía un empleo remunerado. El 43,9% (18; 41) registró comorbilidades: hipotiroidismo 12,20% (5; 41), hipertensión arterial 9,76% (4; 41) y diabetes 4,88% (2; 41), el 12,20% (5; 41) recibieron tratamiento para depresión y 4,88% (2; 41) mantuvieron seguimiento por gastritis crónica. La asociación con otras enfermedades autoinmunes se evidenció en 12,20% (5; 41), que tuvieron diagnóstico de tiroiditis de Hashimoto 7,32% (3; 41) y síndrome de Sjogren 4,88% (2; 41).
El promedio de edad al inicio de los síntomas fue 41,12 años (DE +/- 15). El 43,9% (18; 41) debutó con neuritis óptica; el 31,7% (13; 41) pacientes presentaron mielitis como primer síntoma y el 24,4% (10; 41) la combinación de neuritis óptica y mielitis fueron los síntomas iniciales. En promedio, el diagnóstico definitivo de NMO demoró 4,12 años (DE +/- 5,5) desde el inicio de los síntomas y la media de duración de la enfermedad fue de 6,8 años (DE +/ -6). El promedio del tiempo de diagnóstico fue 3,17 años (DE +/- 1,59). El 87,8% (36; 41) con un fenotipo recurrente y tuvieron un promedio de 3,7 brotes en total (DE +/- 2,5). En el 70,7% (29; 41), se identificaron anticuerpos anti- AQP4 en suero mediante inmunofluorescencia directa, el 4,9% (2; 41) no se realizaron esta prueba diagnóstica.
La media del puntaje de discapacidad fue 4,54 puntos (DE +/- 1,68), el paciente con menor discapacidad tuvo 2 puntos y el caso con mayor limitación tuvo 8 puntos. El promedio de puntuación en la escala funcional visual fue 3 puntos (DE +/- 1,2) y en la escala funcional piramidal fue 2,73 puntos (DE +/- 1,55). El 51,2% (21; 41) requirieron un apoyo uni o bilateral para la marcha.
El 73,2% (30; 41) recibió Rituximab (RTX) como tratamiento de mantenimiento, 9,8% (4; 41) eran tratados con Micofenolato y el 7,3% (3; 41) se mantuvo con Azatioprina. El 9,8% (4; 41) recibieron tratamiento combinado. Fallecieron 9,8% (4; 41), con un promedio de edad de 43 años (DE +/- 10). Los decesos en relación a causas infecciosas como shock séptico fue el 100,0%; de origen pulmonar 7,3% (3; 41) y gastrointestinal 2,4% (1; 41), ninguna de las muertes ocurrió en la institución.
Se conoció que la enfermedad es frecuente en regiones donde predominan etnias no caucásicas y la prevalencia de EM fue baja en regiones cercanas a la línea ecuatorial6,8. En Ecuador no se conoce la prevalencia exacta de la enfermedad, sin embargo, el último censo nacional realizado en el año 2010 reportó que el 71,9% de la población era mestiza, lo que sumada a la su situación geográfica, podrían traducir en una mayor frecuencia de la enfermedad9,10. Alvarenga et al., se incluyeron países donde la población blanca y de origen caucásico fue más prevalente; en relación a la situación epidemiológica de la NMO en Latinoamérica encontró una afectación del 45,5% en blancos; 39,9% en afro descendientes; 13,3% en mestizos; el 1,0% en asiáticos y el 1,3% en otras etnias11; datos diferentes al estudio del hospital en el que el 100,0% fueron mestizos.
Al comparar la distribución geográfica de los pacientes con las tasas calculadas de prevalencia de EM en el país, el mayor número de casos de NMO provinieron de provincias con alta proporción de población mestiza e indígena, en Pichincha el 82,1% de la población se connotó mestiza12,13. El estudio en esta Casa de Salud fue limitado por ser local y no contempló la prevalencia, sin embargo, dio una idea del comportamiento de la enfermedad en relación a las etnias.
La NMO es común en mujeres con una relación 5-10:1 a hombres, se sugirió la posibilidad de una influencia hormonal en la fisiopatología de la enfermedad, sin lograr establecer el mecanismo1,14. Puede ocurrir a cualquier edad, afecta de manera principal a poblaciones con edades comprendidas entre los 30 y 40 años6,10. Los resultados concordaron con esta epidemiología por el claro predominio de mujeres (relación 3:1) y el promedio de edad de los pacientes al inicio de los síntomas fue de 41,1 años, fue la causa de discapacidad en adultos jóvenes, en edad productiva como se evidenció en los datos donde el 65,9% de pacientes mantuvieron un empleo remunerado. Su presentación en edades pediátricas o mayores de 65 años correspondió al 15-20% de todos los pacientes con NMO2,15 pero no se incluyeron casos pediátricos en el estudio.
Un estudio retrospectivo observó que la hipertensión arterial fue la comorbilidad más común en los pacientes con NMO, lo que se asemejó con la descripción del estudio en éste nosocomio16. La asociación con otras entidades autoinmunes se ha observado hasta en un 30,0% de pacientes, siendo las patologías más comunes: la Miastenia Gravis, Síndrome de Sjögren y Lupus Eritematoso Sistémico2,15,17,18. Zhong et al., analizaron la relación entre NMO y la coexistencia de Síndrome de Sjögren, sin encontrar un curso más activo de la enfermedad o lesiones severas en el SNC, ni empeoramiento del EDSS en estos19. Cinco de los pacientes recibieron tratamiento para la depresión, misma que se vio asociada a NMO en la literatura debido al deterioro global que presentan en el curso de la enfermedad. Un estudio alemán mostró que el 28,0% padecieron depresión moderada - severa y en el 48,0% se relacionaba a dolor neuropático, pero el 40,0% de todos recibió tratamiento médico antidepresivo20. Las ideas suicidas también han sido reportadas, aunque este dato no se estudió1.
Se conoce que hasta el 80,0% de los pacientes presentaron un patrón recurrente, caracterizado por ataques severos que provocan déficits neurológicos permanentes. En el 20,0% de casos se puede presentar como un único evento de neuritis óptica o mielitis extensa sin evidencia de nuevos ataques, lo que se consideró un curso monofásico; y el de tipo progresivo secundario fue raro6,18. Este comportamiento de la enfermedad se cumplió en el grupo donde la mayoría tuvo un fenotipo recurrente.
Carnero et al., en su estudio sobre NMO en Latinoamérica, evidenciaron que la neuritis óptica sola o concomitante con mielitis transversa (56,7%) y mielitis transversa aguda (45,1%) fueron los síntomas frecuentes al inicio de la enfermedad21, lo que coincidió con la descripción de neuritis óptica (43,9%) y mielitis (31,7%). Otros estudios reportaron frecuencias similares de síntomas para neuritis óptica (33-45%), mielitis transversa (37-61%), síndrome de área postrema (7- 16%), síndrome del tronco encefálico (8- 31%), síndrome diencefálico (15,0%) y síndrome cerebral (40-60%) que incluyó encefalopatía, convulsiones y hemiparesia en pacientes con NMO7,22-26.
Al hablar de anticuerpos anti-AQP4, los datos concordaron con estudios realizados en países latinoamericanos como Colombia y Costa Rica donde se describió la presencia de anticuerpos anti-AQP4 positivos en el 33-74% de pacientes10,11,27. El fenotipo recurrente fue común en mujeres y una mayor asociación con la positividad de anti-AQP4, como los hallazgos encontrados14,15. En éste estudio no se conoció la prevalencia de positividad de anticuerpos contra la glicoproteína de la mielina de oligodendrocitos (anti-MOG) porque no se realizó el test, lo que representó una limitación. Se ha descrito que el 20-25% de los pacientes seronegativos para anticuerpos anti-AQP4 pueden ser positivos para anticuerpos anti-MOG2,18.
Los hallazgos en discapacidad fueron similares a los encontrados en un estudio colombiano del año 2016 donde los pacientes mostraron una mediana de EDSS de 4.1 puntos27. En poblaciones sudamericanas reportaron que la discapacidad fue leve en el 25,7%, moderada del 41,2% y la severa fue 32,3%11. Esta descripción no concordó con lo observado. En el estudio, el 51,2% requirieron un auxiliar para la marcha ya sea unilateral o bilateral (discapacidad severa).
La mayoría de pacientes recibieron RTX como tratamiento de mantenimiento y no existe un fármaco específico para el manejo de esta enfermedad. Tres ensayos clínicos controlados randomizados demostraron que los nuevos anticuerpos monoclonales (Inebilizumab, Eculizumab y Satralizumab) redujeron las recaídas en pacientes con NMO entre 62-94,2% en comparación con el grupo placebo28-30. Ninguno de estos fármacos estuvo disponible en el país para el tratamiento de pacientes con NMO. Varios fármacos inmunosupresores como Azatioprina (AZA), Micofenolato Mofetilo (MMF) y RTX, han sido utilizados como terapias a largo plazo para NMO tomando en cuenta los mecanismos fisiopatológicos31. La evidencia, consideró a los linfocitos B como un blanco terapéutico en el tratamiento de la NMO, por lo que se propuso el uso de un anticuerpo monoclonal anti CD20 como tratamiento de primera línea32,33.
Otra limitación del estudio fue ser retrospectivo, en un grupo pequeño de pacientes y en un solo centro de referencia. Sin embargo, la recolección de datos sobre las características de los pacientes fue detallada y confiable al contar con registros clínicos digitales. Este estudio puede ser la base para la replicación en otros hospitales.
Se determinó el perfil clínico y epidemiológico de los pacientes con diagnóstico de trastorno del espectro de neuromielitis óptica. Los resultados mostraron similitud con otros estudios sobre el comportamiento clínico y epidemiológico de la enfermedad. Existió demora en el diagnóstico definitivo de los pacientes desde el inicio de los síntomas, lo que se tradujo en un aumento de la discapacidad. Se determinó la necesidad de mejorar el diagnóstico y tratamiento inmediato que evite la progresión de la discapacidad en los pacientes.
AQP4: Anticuerpo anti acuaporina 4; AZA: Azatioprina; CIE: Clasificación Internacional de Enfermedades; DE: Desviación estándar; EDSS: Escala expandida de discapacidad; EM: Esclerosis Múltiple; IBM SPSS: International Business Machines Statistical Package for the Social Sciences; MMF: Micofenolato mofetilo; MOG: Glicoproteína de la mielina de oligodendrocitos; NMO: Neuromielitis Óptica; RTX: Rituximab; SNC: Sistema nervioso central.
FC: Obtención de resultados, asesoría estadística, asesoría técnica o administrativa. WG: Obtención de resultados. JM: Recolección y obtención de resultados. EC, FC, WG, JM: Concepción y diseño del trabajo, Análisis e interpretación de datos, Redacción del manuscrito, Revisión crítica del manuscrito, Aprobación de su versión final y Rendición de cuentas.
Edgar Patricio Correa Díaz. Médico, Diploma Superior en Educación en Ciencias de la Salud, Universidad Central del Ecuador. Especialista en Neurología, Universidad San Francisco de Quito. Jefe de la Unidad de Neurología, Hospital de Especialidades Carlos Andrade Marín. Docente, Facultad de Medicina, Pontificia Universidad Católica del Ecuador. Quito-Ecuador. ORCID: https://orcid.org/0000-0001-6363-8886
Francisco José Caiza Zambrano. Médico Cirujano, Pontificia Universidad Católica del Ecuador. Médico General en Funciones Hospitalarias, Unidad de Neurología, Hospital de Especialidades Carlos Andrade Marín. Quito-Ecuador. ORCID: https://orcid.org/0000-0001-6902-1545
Wilson Alfredo Gualotuña Pachacama. Médico, Universidad Central del Ecuador. Médico General en Funciones Hospitalarias, Unidad de Neurología, Hospital de Especialidades Carlos Andrade Marín. Quito-Ecuador. ORCID: https://orcid.org/0000-0001-7256-8795
Joselyn Elizabeth Miño Zambrano. Médica, Universidad Central del Ecuador. Médico General en Funciones Hospitalarias, Unidad de Neurología, Hospital de Especialidades Carlos Andrade Marín. Quito-Ecuador. ORCID: https://orcid.org/0000-0002-7483-3481
Se utilizaron recursos bibliográficos de uso libre y limitado. La información recolectada está disponible bajo requisición al autor principal.
El estudio fue aprobado por pares y por el Comité de Ética de Investigación en Seres Humanos–CEISH/HCAM.
La publicación fue aprobada por el Comité de Política Editorial de la Revista Médica Científica CAMbios del HECAM en Acta 002 de fecha 20 de mayo de 2021.
Estudio financiado con fondos propios de los autores.
Los autores declaran no tener ningún conflicto de interés personal, financiero, intelectual, económico y de interés corporativo.