Impacto cardiovascular, conducta diagnóstica y terapéutica del feocromocitoma como causa de hipertensión arterial.
Impacto cardiovascular, conducta diagnóstica y terapéutica del feocromocitoma como causa de hipertensión arterial. |
Impacto cardiovascular, conducta diagnóstica y terapéutica del feocromocitoma como causa de hipertensión arterial.
Cardiovascular impact, diagnostic and therapeutic management of pheochromocytoma as a cause of arterial hypertension.
Mariela Viviana Villagómez Estrada1, Jorge Luis Salazar Vega2
INTRODUCCIÓN. Los feocromocitomas son tumores que provienen de las células neuroendócrinas
de la médula adrenal y producen alta secreción de catecolaminas. Generan complicaciones
cardiovasculares graves que suelen asociarse con crisis hipertensivas. Es importante
valorar el impacto cardiovascular de esta entidad. OBJETIVO. Realizar una revisión exhaustiva
de las diversas manifestaciones de los feocromocitomas como causa de hipertensión arterial,
su impacto cardiovascular, conducta diagnóstica y terapéutica. MATERIALES Y MÉTODOS.
Revisión bibliográfica y análisis de 141 artículos científicos que incluyeron temas sobre el impacto
cardiovascular, conducta diagnóstica y terapéutica del feocromocitoma como causa de
hipertensión arterial. Se usó bases de datos: Medline, Embase, Scopus, Pubmed, Google Académico.
Criterios de búsqueda en DECS, MeSH: “pheochromocytoma OR hypertension arterial
AND cardiomyopathy”, en inglés- español. Fueron seleccionados: 13 publicaciones de texto
completo, 10 artículos retrospectivos, 2 guías de práctica clínica y 1 revisión. Se excluyeron
128 artículos científicos. RESULTADOS. Se realizó una revisión de las manifestaciones clínicas
de los feocromocitomas como causa de hipertensión arterial y el impacto cardiovascular
se relacionó con la producción de catecolaminas. Para el diagnóstico, la sensibilidad de la
resonancia magnética es del 93-100%; la especificidad de resonancia magnética o tomografía
computarizada en combinación con gammagrafía con metayodobencilguanidina con 123I es
cercana al 100%. La resección del feocromocitoma tiene potencial curativo. CONCLUSIÓN. Los
feocromocitomas presentan variabilidad clínica, se asocian a complicaciones cardiovasculares
y cerebrovasculares graves por producción de catecolaminas. El diagnóstico oportuno y eficaz
debe realizarse mediante resonancia magnética y gammagrafía en caso de alta sospecha clínica.
El tratamiento quirúrgico es de elección.
Palabras clave:Feocromocitoma/diagnóstico; Cardiomiopatías; Hipertensión; Catecolaminas/
uso terapéutico; Neoplasias de las Glándulas Suprarrenales/cirugía; Enfermedades del Sistema
Endócrino.
INTRODUCTION. Pheochromocytomas are tumors arising from the neuroendocrine cells of
the adrenal medulla and produce high secretion of catecholamines. They generate severe cardiovascular
complications that are often associated with hypertensive crises. It is important to
assess the cardiovascular impact of this entity. OBJECTIVE. To perform an exhaustive review
of the various manifestations of pheochromocytomas as a cause of arterial hypertension, their
cardiovascular impact, diagnostic and therapeutic conduct. MATERIALS AND METHODS. Bibliographic
review and analysis of 141 scientific articles that included topics on the cardiovascular
impact, diagnostic and therapeutic behavior of pheochromocytoma as a cause of arterial
hypertension. The following databases were used: Medline, Embase, Scopus, Pubmed, Google
Scholar. Search criteria in DECS, MeSH: “pheochromocytoma OR hypertension arterial AND
cardiomyopathy”, in English-Spanish. The following were selected: 13 full-text publications, 10
retrospective articles, 2 clinical practice guidelines, and 1 review. A total of 128 scientific articles
were excluded. RESULTS. A review of the clinical manifestations of pheochromocytomas
as a cause of arterial hypertension was performed and the cardiovascular impact was related
to catecholamine production. For diagnosis, the sensitivity of MRI is 93-100%; the specificity
of MRI or computed tomography in combination with 123I-methiodobenzylguanidine scintigraphy
is close to 100%. Resection of pheochromocytoma has curative potential. CONCLUSION.
Pheochromocytomas present clinical variability, are associated with severe cardiovascular and
cerebrovascular complications due to catecholamine production. Timely and effective diagnosis
should be made by MRI and scintigraphy in case of high clinical suspicion. Surgical treatment is
the treatment of choice.
Keywords: Pheochromocytoma/diagnosis; Cardiomyopathies; Hypertension; Catecholamines/
therapeutic use; Adrenal Gland Neoplasms/surgery; Endocrine System Diseases.
Los feocromocitomas (FCT) son tumores que provienen de las células neuroendocrinas de la médula adrenal, capaces de producir y secretar catecolaminas (adrenalina, noradrenalina y dopamina). Neoplasias con similar capacidad secretora (noradrenalina y dopamina) pueden provenir de los ganglios simpáticos paravertebrales y se denominan paragangliomas (PGL) o FCTs extraadrenales1,2. Se ha reportado una incidencia anual de FCTs y PGLs (FPGLs) entre 0,4 a 9,5 casos por millón de habitantes1 con una prevalencia entre 1:2 500 y 1:6 5003,4.
El 5-10% de los pacientes con hipertensión arterial (HTA) puede identificarse como HTA secundaria5. La HTA endocrina es la segunda causa común de HTA secundaria después de la enfermedad renal crónica. En 0,1 a 1,0% de personas con HTA puede reconocerse FPGLs6,7.
La presentación de los FCTs es heterogénea. Entre 10-49% son detectados como incidentalomas4. En otros escenarios pueden detectarse FCTs por cuadros clínicos atribuibles a la hipersecreción de catecolaminas, observables en forma crónica o paroxística8. El FCTs ha sido denominado “el gran simulador” desde su descripción inicial, pues imita más de 30 patologías médicas9,10.
Las complicaciones cardiovasculares graves en los FPGLs suelen estar asociadas con crisis hipertensivas. En estudios retrospectivos se ha demostrado una incidencia relativamente alta de complicaciones cardiovasculares que varía entre el 19,3-36,0% de pacientes con FCTs8,11. Entre ellas se identifican: isquemia miocárdica, arritmias, disfunción miocárdica (con o sin insuficiencia cardíaca), hemorragia cerebral y muerte súbita12.
Cuando se evaluaron post mortem las causas de defunción en personas con FCTs no diagnosticados y no tratados, se detectó que el 71,0% fallecieron por etiologías cardiovasculares, como infarto de miocardio (IM) e insuficiencia cardíaca13.
Ante la sospecha clínica se requiere la realización de estudios bioquímicos y de imagen para confirmar y localizar la enfermedad. La resección quirúrgica es el tratamiento de elección. Se debe realizar una adecuada preparación farmacológica para minimizar las complicaciones asociadas a la cirugía2.
Los FPGLs son una patología compleja, de baja prevalencia, con elevado riesgo de mortalidad y complicaciones cardiovasculares, cuyo manejo deseable es multidisciplinario y en unidades especializadas14. El objetivo de ésta revisión fue analizar las características clínicas (con especial enfoque en las complicaciones cardiovasculares), la evaluación diagnóstica y medidas terapéuticas en personas afectadas por FCTs, al recopilar y analizar información que permitirá un óptimo manejo de los pacientes.
Se realizó una revisión bibliográfica y análisis de 141 artículos científicos que incluyeron temas sobre el impacto cardiovascular, conducta diagnóstica y terapéutica del feocromocitoma como causa de hipertensión arterial, encontrada en las bases de datos: Medline, Embase, Scopus, Pubmed, Google Académico. Los artículos fueron: estudios en idioma inglés-español que cumplieron criterios de búsqueda: DECS, MeSH: “pheochromocytoma OR paraganglioma OR hypertension arterial AND cardiomyopathy”; se filtró los datos obtenidos mediante los parámetros “full text”, “retrospectivos” y “guías de práctica clínica”. Fueron seleccionados: 13 publicaciones de texto completo, 10 artículos retrospectivos, 2 guías de práctica clínica y 1 revisión. Se excluyeron 120 artículos científicos que no registraron información sobre el impacto cardiovascular de feocromocitoma; 3 por ser reportes de caso con un participante y 5 por ser comentarios. Se limitó la búsqueda a literatura de los últimos 20 años.
La revisión de la evidencia científica determinó:
La hipersecreción episódica hormonal puede resultar en una tríada clásica de síntomas: dolor de cabeza, sudoración y palpitaciones, reportada en un aproximado del 25% de los pacientes con FCTs15. Esta tríada junto a HTA de manera concomitante tiene una especificidad diagnóstica superior al 90%16,17. En esta revisión analizamos 4 estudios retrospectivos, donde el 15,71% (58; 369) de pacientes presentaron la triada clásica6,8,18. Tabla 1.

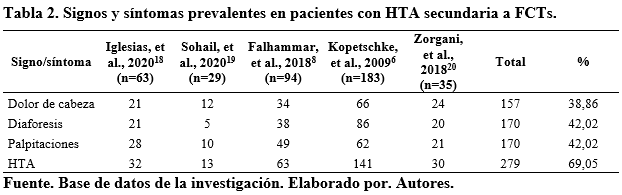
Otros síntomas pueden incluir palidez, náuseas, vómitos, estreñimiento, rubor, pérdida de peso, debilidad, fiebre, hipotensión ortostática, dolor torácico o abdominal y muerte súbita. También se han referido trastornos psiquiátricos como: ataques de pánico, deterioro cognitivo reversible, trastornos de ansiedad (20-40% de los casos) y depresión (esta última descrita en particular en personas con neoplasia endocrina múltiple tipo 215,20-23. En la tabla 2 se muestran las manifestaciones más frecuentes, se analizaron 5 estudios con un total de 404 pacientes.
Todos estos síntomas pueden ocurrir de forma aislada y, dado su carácter inespecífico, el diagnóstico a veces es un desafío24.
Los efectos de la HTA prolongada pueden precipitar el daño de órganos a nivel: cardíaco, renal, ocular, del sistema nervioso central y desregulación del metabolismo glucémico llegando a causar hiperglucemia y diabetes25-27.
FPGLs e hipertensión arterial
Los FPGLs se notifican en el 0,1-1% de
los pacientes hipertensos6,7, a su vez la
HTA es una manifestación muy común
que puede ocurrir hasta en un 95% de
los pacientes con FPGL17. Se ha informado
en un estudio retrospectivo que
los pacientes con FCTs tienen una tasa
de eventos cardiovasculares 14 veces
mayor que aquellos con HTA28.
La hipersecreción de catecolaminas puede ser continua o intermitente, resultando en una presión arterial fluctuante con episodios cíclicos que abarcan: HTA sostenida o crónica, HTA paroxística, HTA sostenida con paroxismos, normotensión e incluso hipotensión ortostática17,29,30. Esta amplia variación clínica está relacionada con el predominio de secreción catecolaminégica tumoral y en función de ello expresión de fenotipos diversos30.
La HTA prolongada y no detectada en los pacientes con FPGLs puede desencadenar miocardiopatía hipertrófica, con características clínicas y ecocardiográficas similares a las de la miocardiopatía hipertrófica (MCH) obstructiva, sin embargo, puede mejorar e incluso revertirse después de la resección del FPGLs31,32.
En un porcentaje variable de pacientes (39-94%) el tratamiento quirúrgico de los FCTs sintomáticos conlleva a una regresión de la HTA, o una reducción de la cantidad o dosis de medicamentos usados en su tratamiento26,33,34.
Crisis por FCTs
En los pacientes con FPGLs el aumento
masivo de catecolaminas puede resultar
en una “crisis por FCTs” que conduce a
una disfunción multiorgánica progresiva.
Tiene una incidencia del 3,02-18%35,36,
con una tasa de mortalidad significativa
que oscila entre 11-16,7%35,37,38.
Esta crisis mortal en potencia, se caracteriza por HTA grave con inestabilidad hemodinámica (colapso cardiovascular) y el compromiso varios sistemas: respiratorio, neurológico, gastrointestinal, renal, hepático y metabólico25,29,39.
Los síntomas abruptos e inespecíficos de la crisis por FCTs plantean desafíos diagnósticos. La variabilidad clínica suele dar lugar a la sospecha de otras entidades, con mayor frecuencia sepsis35. El retraso en su identificación pospone la aplicación de un tratamiento adecuado y empora el pronóstico. En consecuencia, cualquier paciente que presente un shock inexplicable o insuficiencia ventricular izquierda, falla multiorgánica, crisis hipertensiva o acidosis láctica inexplicable, deber ser candidato para el diagnóstico de crisis por feocromocitoma35,40.
Dentro de los desencadenantes se incluyen: estrés psicológico intenso (ansiedad, dolor severo o esfuerzo), estrés mecánico (coito, defecación o palpación del tumor), inducción anestésica o la intubación, embarazo (movimientos fetales, contracciones uterinas, parto normal o cesárea), además de ciertos fármacos (glucocorticoides, antagonistas del receptor de dopamina, bloqueadores de los receptores beta-adrenérgicos, opioides, simpaticomiméticos, inhibidores de la receptación de noradrenalina/ serotonina, etcétera)35. Además, existen reportes de crisis por feocromocitoma de aparición espontánea sin ningún estrés exógeno8,17.
Arritmias
Se encuentran en el 20% de los pacientes
con FCTs. El síntoma clínico más importante
de arritmia en los FPGLs son las
palpitaciones32. En un estudio actual de
revisión de electrocardiogramas obtenidos
en 650 pacientes con FCTs, se encontró
que el 10,9% mostró arritmias.
Las alteraciones más frecuentes fueron:
taquicardia sinusal 98,6%, fibrilación auricular
11,3%, aleteo auricular 5,6%; y taquicardia
ventricular 4,2%41. El bloqueo
auriculoventricular es una complicación
poco común32. En la revisión citada ninguno
de los pacientes tuvo recurrencia de
arritmias después del tratamiento quirúrgico
y/o farmacológico apropiado41. La
estrategia de tratamiento en las taquiarritmias
inducidas por catecolaminas se
basa en gran medida en el bloqueo de los
receptores adrenérgicos beta 1 y también
de calcio antagonistas41,42.
Síndrome coronario agudo
El exceso de catecolaminas induce vasoconstricción
y vasoespasmo de las arterias
coronarias. Esto puede resultar en
isquemia miocárdica y un potencial infarto,
en ausencia de enfermedad coronaria
aterosclerótica significativa43. Las
catecolaminas alteran la permeabilidad
de la membrana sarcolémica y ocasionan
cambios en el flujo del calcio intracelular,
así como un proceso inflamatorio difuso
y necrosis miocárdica44. Un número importante
de pacientes con FPGLs presentaron
síndrome coronario agudo. La
mayoría de estos pacientes tenían arterias
coronarias permeables, la histología
del miocardio se interpretó como “daño
miocárdico extenso con infarto e infiltración
de células polimórficas”. Esto
se considera un ejemplo de insuficiencia
coronaria inducida por el metabolismo,
debido a un desequilibrio entre la oferta
y la demanda causados por una estimulación
adrenérgica intensa32. Además, se
ha descrito el IM sin aterosclerosis coronaria
obstructiva (MINOCA)45.
Miocardiopatia
La miocardiopatía inducida por catecolaminas
en feocromocitomas (MCF) es
una complicación rara pero peligrosa.
Una vez que se diagnostica esta afección,
el pronóstico es malo y el riesgo
quirúrgico es mayor. Los estudios retrospectivos
informaron una prevalencia de
MCF de 8-11%44.
La estimulación excesiva inducida por catecolaminas a los miocitos cardíacos conduce a daños estructurales. La exposición crónica a las catecolaminas resulta en fibrosis, apoptosis de los miocitos y reducción de la contractibilidad que se manifiesta en varias formas de miocardiopatía que va desde miocardiopatía de Takotsubo (MT) hasta miocardiopatía dilatada (MCD). Figura 1.
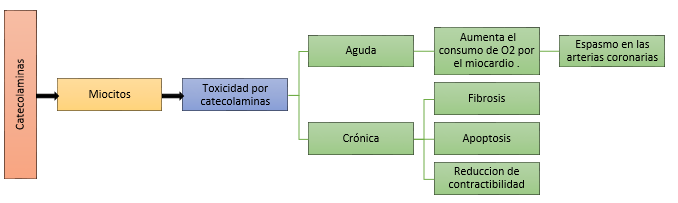
En un análisis reciente sobre la asociación de FCTs y MCF46 se incluyeron 163 casos que presentaron: MCD 63 personas, MT en 68, MCH en 10, miocarditis en 8 y de tipo no especificado 14. Se desconoce la prevalencia de MT en FPGLs, sin embargo, Giavarini, et al., estudiaron 140 pacientes con FPGLs y encontraron que el 11% sufrió miocardiopatía aguda por catecolaminas48. El MT inducido por FPGLs se caracterizó por una presentación clínica dramática acompañada de una alta tasa de complicaciones intrahospitalarias que se produjeron en el 71,8%, incluido el shock cardiogénico en casi el 40,0% y la muerte en el 3,7%47. Se evidenció recuperación después del tratamiento médico o la resección del tumor49.
Complicaciones cerebro
vasculares
Se han notificado en pacientes con FCTs
en un aproximado del 4,8%50, entre ellos
se destacan eventos isquémicos transitorios,
hemorragia subaracnoidea, deterioro
neurológico difuso debido a lesiones isquémicas
de la sustancia blanca11.
Diagnóstico
Cuando se sospecha de FPGLs, se deben
aplicar pruebas bioquímicas para diagnosticar
o excluir la enfermedad, luego se determinará
la ubicación anatómica del tumor.
Bioquímico: La adrenalina y la noradrenalina son metabolizadas por la catecolamina- O-metiltransferasa en metanefrina y normetanefrina, de manera respectiva (metabolitos inactivos)51. La producción y secreción de catecolaminas por el tumor es a menudo reducida y de carácter episódico, a diferencia de la de sus metabolitos inactivos, cuya producción y secreción se realiza de forma continua2,24, convirtiendo a las metanefrinas/normetanefrinas medidas en un marcador muy sensible y con pocos falsos negativos en el estudio de los feocromocitomas.
No se recomienda la medición de ácido vanillilmandélico (VMA) en orina de 24 horas debido a sus altas tasas de falsa negatividad52. Las muestras de sangre deben tomarse, después que el paciente haya permanecido en decúbito supino durante al menos 30 minutos, para evitar falsos positivos53. Cuando la prueba se realiza estando de pie, la tasa de falsos positivos aumenta 2,8 veces en comparación con la posición supina54.
La Endocrine Society recomienda mediciones de metanefrinas en suero u orina en su guía de práctica, sin sugerencias sobre la superioridad de una sobre la otra2. Sin embargo, un estudio reciente, reveló que a pesar de la baja tasa de falsa negatividad de las mediciones de metanefrina libre tanto en plasma como en orina en el diagnóstico de FPGL, se había demostrado un mayor rendimiento diagnóstico de las mediciones en plasma, considerándola como examen de elección. Además, es más conveniente en cuanto a costos y el cumplimiento es superior en comparación con la recolección de orina de 24 horas55. La especificidad y sensibilidad de la metanefrina en orina es del 94% y 93% de manera respectiva56.
Algunas drogas (beta bloqueadores, antidepresivos tricíclicos, paracetamol, cafeína, levodopa, inhibidores de la monoaminooxidasa (IMAO), y simpaticomiméticos); así como determinados eventos (edema agudo de pulmón, infarto agudo al miocardio) pueden generar falsos positivos porque incrementan las concentraciones de catecolaminas en sangre. Así mismo, los pacientes con enfermedad renal crónica también tienen concentraciones elevadas de metanefrinas53,54.
Imagen: Los métodos de imagen contribuyen a localizar y evaluar la anatomía de la masa tumoral, permitiendo la planificación del abordaje quirúrgico. Tanto la tomografía computarizada (TC) como la resonancia magnética (RM) abdominal son útiles. Sin embargo, se prefiere la TC dada su alta rentabilidad y valor de sensibilidad, logrando detectar FCTs de al menos 0,5 cm de diámetro con una sensibilidad del 85-94% para FCTs y un aproximado de 90% para PGL57.
La RM permite una mejor caracterización del tumor y el entorno circundante, lo que permite la exclusión de la invasión vascular; de manera similar, permite una mejor distinción entre tejidos blandos y, por lo tanto, es superior en su capacidad para diferenciar entre FCTs y adenomas suprarrenales24,57. La sensibilidad diagnóstica de la RM se sitúa entre el 93% y el 100% para FCTs y un aproximado del 90% en casos de PGL, metástasis o recidiva22. Sin embargo, tanto la RM como la TC tienen baja especificidad (70-80%); si se combinan con la gammagrafía con metayodobencilguanidina con (123I-MIBG) la especificidad aumenta cerca al 100%24.
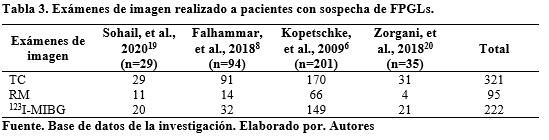
La literatura menciona que la gammagrafía con 123I-MIBG tiene una especificidad diagnóstica en extremo alta del 95-100% y una sensibilidad del 77-90%22. En la presente revisión se evidenció una sensibilidad que varió del 81,90-100%18- 20. Además, se analizaron los exámenes de imagen realizados en 4 estudios con una población de 359 pacientes, se evidenció que el examen realizado con mayor frecuencia es la TC. La sensibilidad y especificidad no se detallan en dichos estudios, por lo que no se mencionan. Tabla 3.
La tomografía por emisión de positrones (PET) es una prueba de imagen alternativa, con nuevos agentes como: 18F-desoxiglucosa (F-FDG), 18F-dihidroxifenalina (F-DOPA) y 18F-fluorodopamina (F-FDA) que se pueden usar en los casos que 123I-MIBG sea negativo y exista una alta sospecha clínica y de laboratorio. En particular, la PET con 18FFDG tiene una mayor sensibilidad que la gammagrafía con 123I-MIBG para la enfermedad metastásica, debido a que, en estos casos, los tumores en general están menos diferenciados y, en consecuencia, pierden su capacidad para captar de manera eficiente la 123I-MIBG24.
En cuanto al diámetro tumoral, en la tabla 4 se muestran las dimensiones analizadas en 5 estudios con un total de 404 pacientes.

Genética: El estudio genético es de suma importancia dada la alta proporción de mutaciones genéticas encontradas en formas esporádicas en apariencia de enfermedad (40%)58 y se recomienda su realización en todos los pacientes con FCTs y PGL2.
La única prueba disponible a nivel nacional es el análisis molecular del proto- oncogen RET59.
Tratamiento
La resección tumoral es el tratamiento de
elección tanto para FCTs como PGL y es la única modalidad terapéutica con potencial
curativo57. La cirugía de un FPGL
es un procedimiento de alto riesgo, la liberación
de cantidades excesivas de catecolaminas,
en especial durante la inducción
anestésica o durante la extirpación
quirúrgica, puede producir complicaciones
cardiovasculares mortales
en potencia, por lo que se requiere un
manejo farmacológico preoperatorio. A
lo largo de los años, la tasa de mortalidad
perioperatoria se ha reducido de manera
significativa hasta 0-3%, debido a la optimización
del tratamiento previo que incluye:
preparación con fármacos, localización
precisa del tumor, introducción
de cirugía mínimamente invasiva y adecuado
manejo de anestesiología60.
En vista de los posibles efectos deletéreos en el sistema cardiovascular, cada paciente con FPGL debe tener un examen preoperatorio completo de la función cardíaca y la presión arterial60.
El objetivo del tratamiento preoperatorio consiste en controlar la HTA y aumentar el volumen circulante. Se requiere realizar de inicio alfa bloqueo y luego beta bloqueo. Está contraindicado el uso inicial de beta bloqueadores, como monoterapia porque la estimulación sin oposición de los receptores alfa puede llevar a vasoconstricción arterial severa53. La fenoxibenzamina es un alfa bloqueador no selectivo de larga acción y se menciona en la literatura como el medicamento de elección, sin embargo su disponibilidad es baja en la mayoría de países, incluido el Ecuador. En la práctica clínica el uso de bloqueadores selectivos alfa 1 de corta acción (doxazosina) es generalizado y con resultados satisfactorios61. En caso de no conseguir el control de cifras tensionales y de frecuencia cardiacas con el bloqueo alfa y beta se pueden adicionar calcio antagonistas o inhibidores de la enzima convertidora de angiotensina. En el postoperatorio se deben vigilar de manera estricta las cifras de tensión arterial y ajustar los medicamentos antihipertensivos para evitar hipotensión53.
Se prefiere el abordaje laparoscópico porque se asocia con estancias hospitalarias más cortas y menores complicaciones postoperatorias. La cirugía abierta es una alternativa, en casos de enfermedad invasiva, para procurar la resección completa del tumor, prevenir su rotura y evitar la recidiva local53.
La adrenalectomía robótica (AR) para el FCT es una opción aceptable con ventajas relativas según datos recientes. Un estudio compara de manera prospectiva los resultados perioperatorios de la adrenalectomía robótica versus la adrenalectomía laparoscópica convencional demostrando que los 2 procedimientos son seguros y eficaces. Los pacientes con niveles altos de normetanefrina pueden beneficiarse de una menor pérdida de sangre y menor tiempo operatorio cuando se utilizó la cirugía robótica, pero la AR tiene un costo mayor62.
En la enfermedad metastásica, el tratamiento se basa en la cirugía de resección tumoral con el objetivo paliativo de reducir el volumen tumoral y la consiguiente reducción de las catecolaminas circulantes, dada su relación directa con la mortalidad y la morbilidad del paciente24.
En casos de enfermedad irresecable y en aquellos en los que la cirugía no es curativa, se puede utilizar quimioterapia y/o radionucleoterapia, en esencia con 131IMIBG63,64. En ausencia de una respuesta de 131I-MIBG insatisfactoria o falta de disponibilidad, se pueden utilizar otras terapias, a saber, octreótido o la quimioterapia, que incluye ciclofosfamida, vincristina y dacarbazina (CVD)65. Sin embargo, aún existe una escasez de estudios controlados que puedan validar la efectividad de estas estrategias en la práctica clínica.
En la enfermedad no metastásica, tanto para FCTs como PGLs, la tasa de supervivencia a cinco años es superior al 95%57. Se conoce que todos los FCTs y PGL tienen algún potencial metastásico66. En comparación con FCTs, los PGL simpáticos presentan un mayor riesgo de metástasis1.
En estos casos, los abordajes terapéuticos son limitados y en su mayoría paliativos, por lo que estos pacientes siempre tienen un mal pronóstico vital.
Las recomendaciones de práctica clínica elaboradas por la Endocrino Society sugieren el seguimiento a largo plazo de todos los pacientes con FCTs o PGLs, independiente del riesgo de recurrencia estimado en un inicio. El control debe ser anual, con perfil bioquímico para determinar la presencia de enfermedad persistente, recurrente o metastásica53.
Los FCTs son entidades infrecuentes con amplia variabilidad de presentación clínica, se asocian a complicaciones cardiovasculares y cerebrovasculares graves por producción de catecolaminas. Se enfatizó la importancia de una alta tasa de sospecha clínica de FCTs para un diagnóstico oportuno y eficaz. El diagnóstico se debe realizar con RM y gammagrafía. El tratamiento quirúrgico es de elección.
Realizar cribado bioquímico con metanefrinas en plasma u orina en todos los pacientes con cuadros sugestivos de FPGL antes de proceder a estudios de imagen para la localización anatómica. Considerar el cribado genético en todos los casos debido a la alta prevalencia de mutaciones en FPGL y realizar seguimiento de por vida.
FCTs: Feocromocitomas; PGLs: Paragangliomas; FPGLs: Feocromocitomas y paragangliomas; HTA: Hipertensión arterial; MINOCA: Infarto de miocardio sin aterosclerosis coronaria obstructiva; MCF: Miocardiopatía inducida por catecolaminas en feocromocitomas; MCD: Miocardiopatía dilatada; MT: Miocardiopatía de Takotsubo; VMA: Ácido vanillilmandélico; TC: Tomografía computarizada; RM: Resonancia magnética; PET: Tomografía por emisión de positrones; F-FDG: 18F-desoxiglucosa; F-DOPA: 18F-dihidroxifenalina; F-FDA: 18F-fluorodopamina; AR: Adrenalectomía robótica; 123I-MIBG: 123I-metayodobencilguanidina; IMAO: Inhibidores de la monoaminooxidasa; PGL: Paraganglioma; IM: Infarto del miocardio; CVD: Ciclofosfamida, vincristina y dacarbazina.
MV, JS: Concepción y diseño del trabajo, análisis e interpretación de datos, redacción del manuscrito. JS: Revisión crítica del manuscrito. Todos los autores leyeron y aprobaron la versión final del artículo científico.
Mariela Viviana Villagómez Estrada. Médico General, Especialista en Medicina Interna, Universidad Central del Ecuador. Tutora de prácticas hospitalarias de pregrado, Universidad San Francisco de Quito. Médico Especialista de Medicina Interna, Hospital Básico Alberto Correa Cornejo. Quito-Ecuador. ORCID ID: https://orcid.org/0000-0001-6838-1469
Jorge Luis Salazar Vega. Doctor en Medicina y Cirugía, Universidad Central del Ecuador. Magister en Fisiopatología Bioquímica y Clínica Endocrinológica, Universidad Austral de Argentina. Médico Especialista en Medicina Interna, Médico Especialista en Endocrinología, Universidad de Buenos Aires. Profesor e investigador en la Universidad de Las Américas, Universidad Central del Ecuador y Pontificia Universidad Católica del Ecuador. Responsable de Endocrinología, Hospital de Especialidades Eugenio Espejo. Quito-Ecuador. ORCID ID: https://orcid.org/0000-0003-0899-4428
Se utilizaron recursos bibliográficos de uso libre y limitado. La información recolectada está disponible bajo requisición al autor principal.
La publicación fue aprobada por el Comité de Política Editorial de la Revista Médico Científica CAMbios del HECAM en Acta 002 de fecha 20 de mayo de 2021.
Se trabajó con recursos propios de los autores.
Los autores reportaron no tener ningún conflicto de interés personal, financiero, intelectual, económico y de interés corporativo.
Agradecemos a: Dra. Rossana Ruiz y Dr. Hugo Miranda, por su soporte en la elaboración y revisión del presente trabajo; así como a colegas y amigos del departamento de Endocrinología del Hospital de Especialidades Eugenio Espejo.