Enfermedad relacionada con IgG4, según criterios ACR/EULAR 2019 sin evidencia de IgG4 en sangre.
Enfermedad relacionada con IgG4, según criterios ACR/EULAR 2019 sin evidencia de IgG4 en sangre. |
Enfermedad relacionada con IgG4, según criterios ACR/EULAR 2019 sin evidencia de IgG4 en sangre.
IgG4-related disease according to ACR/EULAR 2019 criteria without evidence of IgG4 in blood.
Rosa Elena Salazar Ponce1, Jessica Esperanza Pinzón Sosoranga2, Rómulo Abad Villacís Tamayo1, Pola Genoveva Velasteguí Cabezas3, Elba Jakeline Salazar Amaya3.
INTRODUCCIÓN. La enfermedad relacionada con IgG4 es una patología fibroinflamatoria multiorgánica,
de origen desconocido, que simula trastornos malignos, infecciosos e inflamatorios.
Los criterios del American College of Rheumatology y la European League against Rheumatism
2019, son útiles para el diagnóstico diferencial de ésta enfermedad cuando se no se cuenta con
evidencia de inmunoglobulina G4 en sangre. CASO CLÍNICO. Paciente hombre de 45 años
de edad, nacido en Ambato-Ecuador, con ingreso en noviembre del 2017, en emergencias del
Hospital de Especialidades Carlos Andrade Marín, con presencia de tos con hemoptisis leve,
febrícula, astenia, pérdida de peso e hiporexia de dos semanas de evolución. Se realizó múltiples
exámenes, tras observar infiltrados pulmonares intersticiales, con elevación de inmunoglobulina
G en suero, negativas para malignidad; se sospechó de enfermedad relacionada a
inmunoglobulina G4. Se ampliaron los estudios para descartar otras patologías más prevalentes
y cuyo diferencial es primordial. Se inició tratamiento con prednisona y micofenolato con buena
respuesta clínica; durante dos años. DISCUSIÓN. La evidencia científica registró que el hallazgo
más importante en la enfermedad relacionada con inmunoglobulina G4 fue un aumento de
sus niveles séricos. La recurrencia de la enfermedad en un órgano afectado o la aparición de
un nuevo órgano involucrado pudo conducir al diagnóstico en el caso presentado. CONCLUSIÓN.
La enfermedad relacionada con inmunoglobulina G4 al ser una patología heterogénea,
inmunomediada, al simular otras afecciones puede retrasar el diagnóstico; se debe tener una
alta sospecha clínica, si al excluir otros procesos infecciosos, autoinmunes y/o neoplásicos, hay
evidencia de patología fibroesclerosante multiorgánica sin causa establecida.
Palabras clave: Enfermedad relacionada con Inmunoglobulina G4; Enfermedades Autoinmunes;
Enfermedad de Mikulicz; Glucorticoides/uso terapéutico; Insuficiencia Multiorgánica; Enfermedades
Reumáticas/diagnóstico.
INTRODUCTION. IgG4-related disease is a multiorgan fibroinflammatory pathology of unknown
origin that mimics malignant, infectious, and inflammatory disorders. The criteria of the American
College of Rheumatology and the European League against Rheumatism 2019 are useful for the
differential diagnosis of this disease when there is no evidence of immunoglobulin G4 in blood.
CLINICAL CASE. 45-year-old male patient, born in Ambato-Ecuador, with admission in November
2017, in the emergency room of the Hospital de Especialidades Carlos Andrade Marín, with
the presence of cough with mild hemoptysis, fever, asthenia, weight loss and hyporexia of two
weeks of evolution. Multiple tests were performed, after observing interstitial pulmonary infiltrates,
with elevated serum immunoglobulin G, negative for malignancy; immunoglobulin G4-related
disease was suspected. Studies were extended to rule out other more prevalent pathologies
whose differential is paramount. Treatment with prednisone and mycophenolate was started
with good clinical response; for two years. DISCUSSION. The scientific evidence recorded that
the most important finding in immunoglobulin G4-related disease was an increase in its serum
levels. Recurrence of the disease in an affected organ or the appearance of a new involved
organ could have led to the diagnosis in the presented case. CONCLUSION. Immunoglobulin
G4-related disease, being a heterogeneous, immune-mediated pathology, by simulating other
conditions may delay diagnosis; a high clinical suspicion should be maintained if, when other infectious,
autoimmune and/or neoplastic processes are excluded, there is evidence of multiorgan
fibrosclerosing pathology without established cause.
Keywords: Immunoglobulin G4-Related Disease; Autoimmune Diseases; Mikulicz’ Disease;
Glucorticoids/terapeutic use; Multiple Organ Failure; Rheumatic Diseases/diagnosis.
La enfermedad relacionada con IgG4 (ERIgG4) es una patología fibroinflamatoria multiorgánica, de origen desconocido, que simula trastornos malignos, infecciosos e inflamatorios1,2. Fue descrita por primera vez en pacientes con pancreatitis esclerosante que tenían niveles elevados de IgG4+ en suero3. Con el paso del tiempo se evidenció que varias enfermedades tenían en común niveles altos de IgG4, con un hallazgo adicional, la evidencia de infiltración de células plasmáticas positivas para IgG4 en sus tejidos2. Las características histológicas de la enfermedad son: presencia de esclerosis de patrón estoriforme, denso infiltrado linfoplasmocitario, y proporción aumentada de células positivas para IgG4 por inmunohistoquímica respecto a las positivas para IgG. El promedio de esta proporción es una relación de células IgG4+/IgG+ mayor al 40%, pero el criterio varía según el órgano afectado1-3. Al utilizar dichos hallazgos histológicos, se estimó que la prevalencia de la (ER-IgG4) en Japón es de 2,63-10,2 casos por millón de habitantes, con una incidencia de 336-1 300 casos nuevos por año, es más frecuente en hombres en edades comprendidas entre los 50 a 70 años, las lesiones se dan en diversos órganos como: páncreas, conductos biliares, glándulas lagrimales, glándulas salivales, tiroides, pulmones, hígado, riñones, sistema nervioso central y periférico4,5. Tanto la tiroiditis de Riedel (tiroiditis fibrosante) como el tumor de Küttner (aumento de tamaño de glándulas submandibulares con fibrosis), la enfermedad de Ormond (fibrosis retroperitoneal) y la de Mikulicz se clasifican dentro del espectro de (ER-IgG4). Aunque los síntomas clínicos varían respecto del órgano afectado, en algunos pacientes se manifiesta con complicaciones graves, como síntomas de obstrucción o compresión debido a organomegalia, hipertrofia o disfunción orgánica causada por la infiltración celular o fibrosis. El tratamiento se basa en corticoides, drogas modificadoras de la enfermedad y terapia biológica anti CD 206. Esta entidad es considerada poco frecuente y su diagnóstico representa un reto clínico por lo que se considera importante dar a conocer el caso de un paciente con síntomas y signos compatibles con esta patología.
Paciente hombre de 45 años de edad, nacido en Ambato-Ecuador, admitido por emergencias en noviembre del 2017, con presencia de tos con hemoptisis leve, febrícula, astenia, pérdida de peso (10kg) e hiporexia de dos semanas de evolución. Al examen físico se observó edema periorbital con eritema conjuntival y secreción purulenta, parótidas incrementadas de tamaño, murmullo vesicular abolido en base izquierda con crepitantes secos bilaterales. Figura 1.
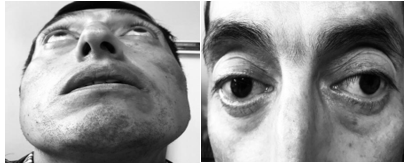
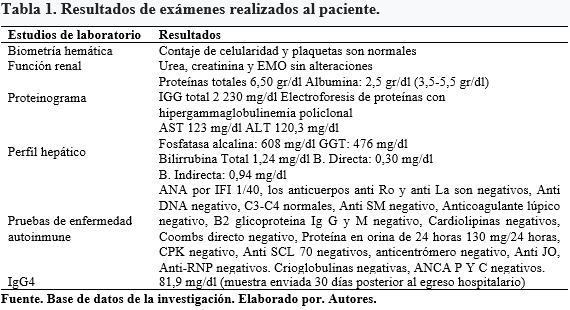
En sus antecedentes personales tiene un largo historial de hospitalizaciones previas, entre los que se destacan una sialoadenitis crónica desde los 25 años de edad, hipertrofia de glándulas lacrimales y episodios de dacriocistitis a repetición que requirió dacriocistorrinostomía a los 40 años de edad, además eritema y edema generalizado palpebral filiado como una celulitis periorbitaria recurrente desde los 42 años, que requirió de tratamiento antibiótico con mejoría parcial e hipertrofia de glándulas parótidas; a los 43 años se diagnosticó de pancreatitis crónica que precipitó insuficiencia pancreática en tratamiento con enzimas pancreáticas e insulina, requirió además de una derivación bilio-digestiva con “Y de Roux” por estenosis benigna en la región distal del colédoco; en el histopatológico se describieron paredes engrosadas y fibróticas de1 cm de espesor y luz de 8 mm, que fue compatible con un proceso inflamatorio crónico de actividad moderada. En el momento del ingreso al Hospital de Especialidades Carlos Andrade Marín (HECAM), se realizaron varios estudios de laboratorio. Tabla 1.
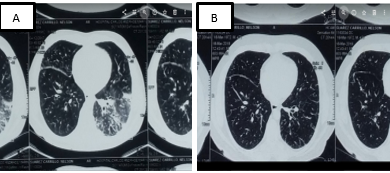
En la tomografía axial computarizada de alta resolución (TACAR) de tórax, se observaron áreas de consolidación, engrosamiento septal, bronquiectasias y ganglios mediastinales menores a 20 mm en nivel 4, 5 y 6. Figura 2.
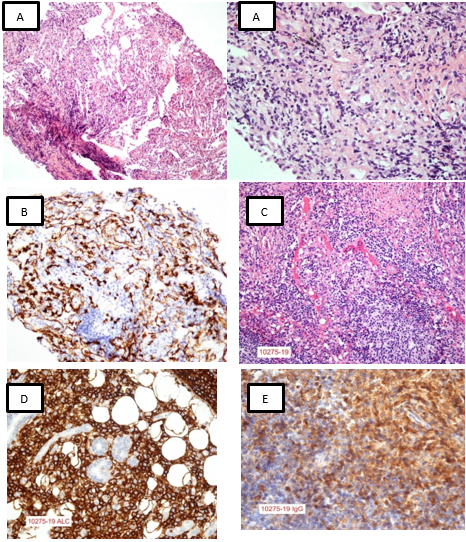
La broncoscopia fue normal, se realizó lavado bronquioalveolar, cepillado y biopsia transbronquial del lóbulo inferior izquierdo; el histopatológico mostró intersticio engrosado e infiltrado linfoplasmocitario denso CD138 positivo en células plasmáticas, tricrómico positivo en proliferación de fibras colágenas en el intersticio pulmonar. No fue posible realizar el contaje de células plasmáticas IgG4/IgG por falta de reactivo. Los cultivos bacteriológicos, micótico y para micobacterias fueron negativos, se complementó con reacción de cadena de la polimerasa (PCR) para micobacterias que también fue negativo. La gammagrafía ósea fue normal. En la tomografía de abdomen se describió un páncreas homogéneo, adelgazado, hígado normal; adrenales, riñones y bazo sin alteraciones, no se observan adenopatías retroperitoneales ni inguinales. Por compromiso poliglandular se solicitó biopsia de parótida en la que se reportó celularidad alc; kappa; lambda: positivos, CD68; CD138: positivo difuso BCl6; CD34; S100; panq: negativos, compatible con reemplazo completo de glándula por infiltrado linfoplasmocitario. Figura 3.
Es un paciente con compromiso inflamatorio crónico de glándulas lacrimales, salivales, parótidas, páncreas y vías biliares con períodos de exacerbación. En la última hospitalización se observaron infiltrados pulmonares intersticiales, con elevación de IgG en suero, con muestras negativas para malignidad; se sospechó de enfermedad relacionada a IgG4. Se ampliaron los estudios para descartar otras patologías más prevalentes y cuyo diferencial es primordial, como infecciones crónicas a micobacterias, hongos, sífilis; neoplasias asociadas a células plasmáticas, linfoma, adenocarcinoma pulmonar; síndrome de Sjogren, vasculitis granulomatosas, sarcoidosis. Tabla 2.

Se aplicaron los criterios de exclusión de otras patologías que simulan enfermedad por IgG4 (tabla 3), se determinó que los dominios que puntúan para enfermedad por IgG4 son: intenso infiltrado linfoplasmocitario, hipertrofia de glándulas lacrimales y parótidas, engrosamiento peribroncovascular y septal cuyos puntajes suman 22, se requieren 20 o más puntos para determinar la presencia de esta patología. Tabla 3.

Se decidió el inicio de tratamiento con prednisona a 40 mg por día, con disminución progresiva, tuvo una descompensación simple de diabetes que requirió ingreso a emergencia donde se incrementó la insulinización, por lo que se añadió al tratamiento micofenolato 1 500 mg por día, lo que permitió el destete progresivo de corticoides con buena respuesta clínica; mejoró el compromiso poliglandular, remisión de la dacrioadenitis, regresión del tamaño de glándulas parótidas, requerimiento mínimo de insulina, ha permanecido en tratamiento por dos años, al momento sin esteroides, continuó con micofenolato, en último control tomográfico de tórax se observó regresión de patrón en vidrio deslustrado y consolidación como se muestra en la figura 3.
La presencia de compromiso poliglandular con síntomas constitucionales que no son atribuidos a patologías autoinmunes, infecciosas o tumorales orienta al diagnóstico de una enfermedad relacionada con IgG4; se ha llegado a señalar que la enfermedad de Mikulicz (1892) caracterizada por inflamación simétrica de las glándulas lagrimal, parótida y submandibular, con infiltración masiva de células mononucleares, el tumor de Küttner (1896) denominada como sialoadenitis crónica esclerosante caracterizada por crecimiento tumoral de la glándula submandibular, la tiroiditis de Riedel y la enfermedad de Odmond (fibrosis retroperitoneal) corresponden al amplio espectro clínico de la enfermedad relacionada con IgG44,5. Se suman a estos hallazgos la pancreatitis con hipergamaglobulinemia que se ha señalado como un prototipo de pancreatitis autoinmune (PAI), que se caracterizó por el hallazgo de esclerosis linfoplasmocítica, hallazgo típico de la PAI relacionada con enfermedad por IgG4. El hallazgo histórico más importante en la enfermedad relacionada con IgG4 que fue un aumento de niveles séricos de IgG4 en pacientes japoneses con PAI7. Alrededor del 60–80% de estos pacientes mostraron ictericia obstructiva con colangitis esclerosante (colangitis esclerosante relacionada con IgG4), cuya imagen no varía de la colangitis esclerosante primaria (CEP), cáncer de páncreas y colangiocarcinoma8. Uno de los hallazgos más interesantes fue que la respuesta a los esteroides y el pronóstico de la colangitis esclerosante asociada a la PAI difirieron de los pacientes con CEP, lo que sugiere diferentes afecciones patológicas9. Se sugirió que la PAI es una enfermedad esclerosante sistémica, esto se basó en los hallazgos de que el páncreas y otros órganos afectados tienen fibrosis con abundante infiltración de células plasmáticas IgG4 positivas10. El perfil histológico y clínico adicional de los pacientes con PAI revela dos subtipos distintos, tipo 1 y tipo 2. La PAI tipo 1 se clasifica como una manifestación pancreática de IgG4 y es probable que sea una enfermedad sistémica con un proceso inmunológico anormal. Se piensa que la PAI tipo 2 es una enfermedad pancreática específica con lesión epitelial granulocítica (LEG) y coexistencia ocasional con colitis ulcerosa9,10. Sobre la base de estos hallazgos, los miembros de los Comités de Investigación Japoneses para “Enfermedad Esclerosante Relacionada con IgG4 Sistémica” (presidido por el Profesor Okazaki) e “IgG4-MOLPS” (presidido por el Profesor Umehara), acordaron el término integral “enfermedad relacionada con IgG4 (IgG4-ER)” que incluye estas condiciones, aunque la patogenia y la fisiopatología siguen sin estar claras11. El primer Simposio internacional sobre ER-IgG4 celebrado en Boston (presidido por el Profesor Stone del Hospital General de Massachusetts) respaldó el concepto japonés y propuso nomenclaturas y criterios patológicos para lesiones de órganos individuales12.
Un panel conjunto del Colegio Americano de Reumatología y la Liga Europea Contra el Reumatismo en el 2018, publicaron los primeros criterios de clasificación para la ER-IgG4, trastorno con una especificidad del 99,2% y una sensibilidad del 85,5%, se debe realizar primero, una exclusión de pacientes antes de aplicar los criterios de clasificación13. La presentación de la ERIgG4 suele ser subaguda, con alteraciones orgánicas evidentes durante meses o incluso años antes del diagnóstico, como el caso del paciente descrito. La enfermedad puede progresar de manera intermitente, con mejoras espontáneas (en general temporales) o largas mesetas de quiescencia de la enfermedad en un órgano específico. En sí, la recurrencia de la enfermedad en un órgano que se sabe está afectado o la aparición de un nuevo órgano involucrado puede conducir al diagnóstico14. Predomina en hombres15. En las cohortes realizadas se evidencia que el compromiso de un órgano aislado es la excepción, mas no la regla en ER-IgG4, que se trata en la mayor parte de los casos de una enfermedad multisistémica15,16, que mostró una diferencia en las manifestaciones según el sexo del paciente.
En el presente caso se observó la presentación oftálmica típica que implica inflamación dentro de la región ocular o proptosis franca, causada por agrandamiento de la glándula lagrimal (dacrioadenitis). La proptosis también puede resultar de pseudotumores orbitales y/o por afectación de músculos extraoculares (miositis orbital), menos comunes fueron la escleritis, la enfermedad del conducto nasolagrimal (obstrucción) y la compresión de los nervios periféricos en el área de la órbita, en particular los nervios trigémino e infraorbitario17. Las glándulas salivales mayores y menores estaban comprometidas, con dacrioadenitis y agrandamiento de las glándulas parótidas y submandibulares; el paciente presentó xerostomía que es un acompañante común de ER-IgG4, pero en general es menos severa que en el síndrome de Sjögren y, a diferencia de este último, puede mejorar con el uso de esteroides; la gammagrafía y sialografía son normales en general18. A diferencia de otros tipos de pancreatitis, la ocasionada por ER-IgG4 no se relaciona con la presencia de autoanticuerpos circulantes. Se manifiesta en general con dolor abdominal (32%), ictericia dolorosa obstructiva (33–59%) edema e infiltración de los ductos pancreáticos y biliares inducida por colangitis esclerosante, dolor de espalda (15%), pérdida de peso (15%) e insuficiencia pancreática exocrina o endocrina (39%) con buena respuesta a esteroides como en el caso presentado4,19.
La afectación torácica en la ER-IgG4 incluye al parénquima pulmonar, las vías respiratorias, el mediastino y la pleura20, las manifestaciones clínicas más frecuentes son tos seca, dolor torácico, fiebre, hemoptisis y disnea progresiva21; el paciente acude a emergencias por tos con hemoptoicos y febrículas. La tomografia pulmonar mostro un compromiso intersticial compatible con neumonía intersticial no específica con infiltrados retículo nodular y vidrio deslustrado. En el pulmón el compromiso puede darse de diversas maneras como un pseudo-tumor inflamatorio, neumonía intersticial (en forma de neumonía intersticial aguda, neumonía intersticial no específica, neumonía intersticial usual, neumonía organizada e incluso bronquiolitis obliterante), adenopatías, estenosis traqueobronquiales, derrame pleural e hipertensión pulmonar22. La ER-IgG4 debe considerarse como un diagnóstico diferencial de neumonía intersticial, si bien son pocos los casos publicados, la mayoría corresponde a neumonía intersticial no específica. Hasta un 50% de los casos puede ser asintomáticos al comienzo de la enfermedad22. Para la tomografía se han descrito diferentes tipos de patrones: lesiones nodulares solidas o masas, vidrio esmerilado, compromiso intersticio-alveolar (engrosamiento de septos inter e intralobulillares tipo panal de abejas y del intersticio peribroncovascular) y bronquiectasias21,22. Estas lesiones también pueden revertir con uso de esteroides, en este caso por la asociación de diabetes fue indispensable el uso de otro inmunosupresor (micofenolato) que permitió destete progresivo y regresión del vidrio deslustrado y áreas de consolidación.
Hay que destacar que en el Ecuador no contamos con reactivos para IgG4 para detectar infiltración de esta inmunoglobulina en tejidos; por costos elevados fuera del país no fue posible hacerlo, esto podría llevar a la especulación de que no se podría hablar en este caso de ER-IgG4; se solicitó niveles de IgG4 en sangre con resultado normal, más el IgG4 sérico, que es un marcador modestamente efectivo para el diagnóstico de ER-IgG423. Sin embargo, el tipo de infiltrados inflamatorios a predominio linfoplasmocitario en los tejidos biopsiados y la respuesta al tratamiento con regresión de todos los hallazgos clásicos de IgG4 ha sido evidente, guiadas además por los criterios de exclusión de otras patologías, así como los criterios de IgG4 con sensibilidad y especificidad elevada, que el paciente cumplió por lo que se concluyó que se trata de esta entidad.
El objetivo del manejo en pacientes con enfermedad relacionada con IgG4 es reducir la inflamación, mantener la remisión de la enfermedad y preservar la función de los órganos mientras se minimizan los efectos adversos del tratamiento. Todos los pacientes con enfermedad activa y sintomática relacionada con IgG4 requieren tratamiento24. Los pacientes que son asintomáticos, pero muestran signos de progresión de la enfermedad en órganos vitales también requieren tratamiento25. Los glucocorticoides son la piedra angular del tratamiento para la mayoría de los pacientes con enfermedad relacionada con IgG4, y los pacientes en general responden bien a esta intervención24-26. De hecho, la falta de respuesta a una dosis apropiada de terapia con glucocorticoides constituye un criterio de exclusión del American College of Rheumatology (ACR) / Criterios de clasificación European League against Rheumatism (EULAR) para enfermedades relacionadas con IgG413. Los fármacos antirreumáticos modificadores de la enfermedad (FARME) (azatioprina, leflunomida, ciclofosfamida, micofenolato) se usan con la intención de reducir la toxicidad de los glucocorticoides, aunque hay poca evidencia de su eficacia a ese respecto. El objetivo de la terapia de inducción en muchos centros es descontinuar los glucocorticoides en 3 a 6 meses, pero las estrategias de tratamiento varían de un país a otro27-29.
Para pacientes con enfermedad multiorgánica y altas concentraciones de IgG4 en suero, es improbable que se produzcan remisiones sin ciclos de glucocorticoides lo tan largos como para causar una toxicidad sustancial. La edad media al diagnóstico de la enfermedad relacionada con IgG4 es de un aproximado de 60 años; en consecuencia, los pacientes a menudo tienen comorbilidades (diabetes, obesidad, osteoporosis e hipertensión)27. Estas comorbilidades comprenden contraindicaciones importantes para la terapia prolongada con glucocorticoides, en particular porque la enfermedad relacionada con IgG4 a menudo tiene un efecto sustancial en el páncreas; en estos casos se debe considerar un agente biológico, en particular Rituximab30. La enfermedad es propensa a la recurrencia, su riesgo aumenta con el número de órganos afectados al inicio y la concentración de IgG4 sérica basal, entre otros factores. Es difícil sugerir una estrategia de mantenimiento de remisión que sea aplicable a todos los pacientes. Las prácticas con respecto a las estrategias de mantenimiento de la remisión difieren en cierto grado de un país a otro, pero se pueden individualizar de acuerdo con las características específicas de la enfermedad del paciente, el alcance del daño relacionado con la enfermedad, las comorbilidades de referencia y las respuestas anteriores al tratamiento29,30.
Kamisawa, et al.19, sugirieron una terapia de mantenimiento a largo plazo con dosis bajas de prednisona por hasta 3 años. Esta estrategia es común en muchos países asiáticos, donde los pacientes en general se mantienen con dosis de prednisona que varían de 5 a 10 mg por día26. Por el contrario, los médicos en los países occidentales han recomendado cursos más cortos de prednisona, con bajas dosis de mantenimiento para pacientes con enfermedad refractaria o para aquellos cuya enfermedad recurre poco después del cese del medicamento30.
Se recomienda una dosis inicial de 0,6 mg/kg por día (30-40 mg por día) de prednisona que es la dosis utilizada en Japón para la pancreatitis autoinmune24,26,30, La dosis inicial de prednisona en general se mantiene durante 2 a 4 semanas. En algunos centros, se inicia un tratamiento con prednisona a 40 mg por día y esta dosis se mantiene durante 4 semanas. Después, la dosis de prednisona a menudo se reduce en 5 mg por día durante 1 a 2 semanas28-30. En este caso el paciente presentó buena respuesta a esteroides como terapia de inducción más micofenolato como terapia de mantenimiento, con regresión de todos los hallazgos clínicos, de laboratorio y mejoría de las imágenes de seguimiento.
A finales del 2019 se publicaron los criterios de clasificación de ER-IgG4, en los que se hace una revisión detallada de las características clínicas, imagenológicas, histopatológicas, el compromiso de órganos; que nos permite hacer un enfoque diagnóstico a partir de una exclusión de otras patologías, y fortalece los criterios de inclusión con puntuaciones mayor o igual a 20 para identificar individuos con características de esta patología. Estos criterios se han desarrollado y validado en una gran cohorte de pacientes, y nos permitirán realizar investigaciones clínicas, epidemiológicas y de ciencias básicas encaminados al mejor conocimiento de la enfermedad.
La enfermedad relacionada con IgG4, al ser una patología heterogénea, inmunomediada, se convierte en la gran simuladora de otras afecciones lo que puede retrasar el diagnóstico, por tanto se debe tener una alta sospecha clínica, si luego de haber excluido otros procesos como infecciosos, autoinmunes y neoplásicos, hay evidencia de patología fibroesclerosante multiorgánica sin causa establecida, un tratamiento oportuno permitirá evitar un daño irreversible que puede poner en riesgo la vida del paciente.
IgG4: Inmunoglobulina G4; IgG4+: Presencia de Inmunoglobulina G4; ERIgG4: Enfermedad relacionada con IgG4; TACAR: Tomografía axial computarizada de alta resolución; PCR: Reacción de cadena de la polimerasa; CEP: Colangitis esclerosante primaria; ACR: American College of Rheumatology; FARME: Fármacos antirreumáticos modificadores de la enfermedad; HE: Hematoxilina eosina; IHQ: Inmunohistoquímica; PAI: Pancreatitis autoinmune; LEG: Lesión epitelial granulocítica; EULAR: European League against Rheumatism; HECAM: Hospital de Especialidades Carlos Andrade Marín.
JP, RS: Concepción y diseño del trabajo, redacción del manuscrito. JP, RS, PV, ES: Recolección/obtención de resultados. RV: Análisis e interpretación de datos. Todos los autores leyeron y aprobaron la versión final del artículo.
Rosa Elena Salazar Ponce. Doctor en Medicina y Cirugía, Especialista en Medicina Interna, Universidad Central del Ecuador. Especialista en Reumatología, Universidad de la Sabana. Médica Reumatóloga, Unidad Técnica de Reumatología, Hospital de Especialidades Carlos Andrade Marín. Quito-Ecuador. ORCID ID: https://orcid.org/0000-0003-2834-8116
Jessica Esperanza Pinzón Sosoranga. Médico, Universidad Central del Ecuador. Especialista en Medicina Interna, Pontificia Universidad Católica del Ecuador. Médico Especialista en Medicina Interna, Unidad Técnica de Medicina Interna, Hospital de Especialidades Carlos Andrade Marín. Quito-Ecuador. ORCID ID: https://orcid.org/0000-0002-8000-1972
Rómulo Abad Villacís Tamayo. Doctor en Medicina y Cirugía, Universidad Central del Ecuador. Especialista en Reumatología, Instituto Ucraniano de posgrado médico Harkov. Jefe de la Unidad Técnica de Reumatología, Hospital de Especialidades Carlos Andrade Marin. Quito- Ecuador. ORCID ID: https://orcid.org/0000-0002-3826-4234
Pola Genoveva Velástegui Cabezas. Doctor en Medicina y Cirugía, Universidad Central del Ecuador. Diplomado superior en pedagogía universitaria, Universidad Nacional de Chimborazo. Especialista en Patología, Universidad Internacional del Ecuador. Médico Patólogo Clínico, Unidad Técnica de Anatomía Patológica, Hospital de Especialidades Carlos Andrade Marín. Quito-Ecuador. ORCID ID: https://orcid.org/0000-0003-0177-0317
Elba Jakeline Salazar Amaya. Doctora en Medicina y Cirugía, Universidad Central del Ecuador. Especialista en Patología, Universidad Técnica Particular de Loja. Patóloga, Unidad Técnica de Anatomía Patológica, Hospital de Especialidades Carlos Andrade Marín. Quito-Ecuador. ORCID ID: https://orcid.org/0000-0003-2744-7403
Se utilizaron recursos bibliográficos de uso libre y limitado. La información recolectada está disponible bajo requisición al autor.
El estudio fue aprobado por pares y por el Comité de Ética de Investigación en Seres Humanos CEISH-HCAM.
La publicación fue aprobada por el Comité de Política Editorial de la Revista Médica Científica CAMbios del HECAM en Acta 006 de fecha 28 de diciembre de 2021.
Se trabajó con recursos propios del autor.
Los autores reportaron no tener ningún conflicto de interés personal, financiero, intelectual, económico y de interés corporativo.